
ZYKADIA 150 mg, gélule, boîte de 15 plaquettes thermoformées de 10
Retiré du marché le : 25/11/2022
Dernière révision : 16/02/2022
Taux de TVA : 2.1%
Prix de vente : 4 196,64 €
Taux remboursement SS : 100%
Base remboursement SS : 4 196,64 €
Laboratoire exploitant : NOVARTIS PHARMA
Source :

Zykadia en monothérapie est indiqué en première ligne de traitement du cancer bronchique non à petites cellules (CBNPC) avancé avec réarrangement du gène anaplastic lymphoma kinase (ALK- positif) chez les patients adultes.
Zykadia en monothérapie est indiqué dans le traitement du cancer bronchique non à petites cellules (CBNPC) avancé avec réarrangement du gène anaplastic lymphoma kinase (ALK-positif) chez les patients adultes préalablement traités par crizotinib.
Hypersensibilité au principe actif ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Hépatotoxicité
Des cas d'hépatotoxicité sont survenus chez 1,1 % des patients traités par céritinib dans les études cliniques. Des élévations de grade 3 ou 4 du taux d'ALAT ont été observées chez 25 % des patients. La plupart des cas ont pu être contrôlés en interrompant le traitement et/ou en réduisant la posologie. Peu d'événements ont nécessité l'arrêt du traitement.
Un bilan hépatique (comprenant un dosage d'ALAT, d'ASAT et de la bilirubine totale) doit être réalisé avant l'instauration du traitement, toutes les 2 semaines pendant les trois premiers mois, puis une fois par mois. En cas d'élévation des transaminases, une surveillance plus fréquente des transaminases hépatiques et de la bilirubine totale doit être mise en place si cliniquement indiqué (voir rubriques Posologie et mode d'administration et Effets indésirables). Des précautions particulières doivent être prises lors du traitement de patients présentant une insuffisance hépatique sévère et la posologie doit être adaptée (voir rubrique Posologie et mode d'administration).
L'expérience est limitée chez ces patients et a montré une aggravation de l'affection hépatique sous- jacente (encéphalopathie hépatique) chez 2 patients sur 10 exposés à des doses uniques de 750 mg de céritinib à jeun (voir rubriques Posologie et mode d'administration, Effets indésirables et Propriétés pharmacocinétiques). D'autres facteurs, en dehors du traitement étudié, pourraient avoir eu un impact sur les évènements observés d'encéphalopathie hépatique, cependant la relation entre le traitement étudié et les événements ne peut pas être complètement exclue. Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère ou modérée (voir rubrique Posologie et mode d'administration).
Pneumopathie interstitielle diffuse/Pneumopathie
Des cas de PID/pneumopathie sévères, menaçant le pronostic vital ou d'issue fatale ont été observés chez des patients traités par céritinib lors des études cliniques. Dans la plupart des cas sévères/ cas menaçant le pronostic vital, l'interruption du traitement a conduit à une amélioration ou une résolution des symptômes.
Les patients doivent faire l'objet d'une surveillance afin de déceler tout symptôme pulmonaire évocateur d'une PID/pneumopathie. Les autres causes potentielles de PID/pneumopathie doivent être exclues et le traitement par céritinib doit être arrêté définitivement en cas de diagnostic de PID/pneumopathie d'origine médicamenteuse, quel qu'en soit le grade (voir rubriques Posologie et mode d'administration et Effets indésirables).
Allongement de l'intervalle QT
Un allongement de l'intervalle QTc a été observé chez des patients traités par céritinib dans les études cliniques (voir rubriques Effets indésirables et Propriétés pharmacocinétiques), ce qui peut entraîner un risque accru de tachyarythmie ventriculaire (par exemple, torsade de pointes) ou de mort subite.
L'utilisation de céritinib chez les patients atteints d'un syndrome du QT long congénital doit être évitée. Les bénéfices et les risques potentiels liés au céritinib doivent être pris en compte avant de débuter le traitement chez les patients présentant une bradycardie préexistante (fréquence cardiaque inférieure à 60 battements par minute [bpm]), les patients ayant des antécédents ou des prédispositions à un allongement de l'intervalle QTc, les patients traités par des anti-arythmiques ou par d'autres médicaments connus pour allonger l'intervalle QT et les patients présentant une pathologie cardiaque significative et/ou des troubles électrolytiques préexistants. Une surveillance régulière par ECG et un suivi régulier des électrolytes (du potassium par exemple) sont recommandés chez ces patients. En cas de vomissements, de diarrhée, de déshydratation ou d'altération de la fonction rénale, corriger le déséquilibre en électrolytes lorsque cela est cliniquement justifié. Le traitement par céritinib doit être arrêté définitivement chez les patients présentant un allongement de l'intervalle QTc > 500 ms ou une variation de l'intervalle QTc > 60 ms par rapport à la valeur de référence et des torsades de pointes, une tachycardie ventriculaire polymorphe ou des signes/symptômes d'arythmie grave. Le traitement par céritinib doit être interrompu chez les patients ayant développé un allongement de l'intervalle QTc > 500 ms sur au moins deux ECG distincts, jusqu'au retour à la valeur de référence ou à un intervalle QTc ≤ 480 ms, puis repris en réduisant la dose de 150 mg (voir rubriques Posologie et mode d'administration, Effets indésirables et Propriétés pharmacocinétiques).
Bradycardie
Des cas de bradycardie asymptomatique (fréquence cardiaque inférieure à 60 bpm) ont été observés chez 21 des 925 (2,3 %) patients traités par céritinib dans le cadre des études cliniques.
L'utilisation de céritinib en association avec d'autres agents connus pour entraîner une bradycardie (par exemple, les bêta-bloquants, les antagonistes calciques non dérivés de la dihydropyridine, la clonidine et la digoxine) doit être évitée autant que possible. La fréquence cardiaque et la pression artérielle doivent être surveillées régulièrement. En cas de bradycardie symptomatique ne menaçant pas le pronostic vital, le traitement par céritinib doit être interrompu jusqu'au retour à une bradycardie asymptomatique ou à une fréquence cardiaque supérieure ou égale à 60 bpm, l'utilisation de médicaments concomitants doit être évaluée et la posologie de céritinib réajustée si nécessaire. En cas de bradycardie menaçant le pronostic vital, le traitement par céritinib doit être arrêté définitivement si aucun traitement concomitant favorisant la bradycardie n'est identifié ; en revanche, en cas d'association avec un traitement connu pour entraîner une bradycardie ou une hypotension, le traitement par céritinib doit être interrompu jusqu'au retour à une bradycardie asymptomatique ou à une fréquence cardiaque supérieure ou égale à 60 bpm. S'il est possible d'ajuster la posologie ou d'arrêter le traitement concomitant, le traitement par céritinib doit être repris en réduisant la dose de 150 mg après le retour à une bradycardie asymptomatique ou à une fréquence cardiaque supérieure ou égale à 60 bpm, avec des contrôles fréquents (voir rubriques Posologie et mode d'administration et Effets indésirables).
Effets indésirables gastro-intestinaux
Des diarrhées, des nausées ou des vomissements sont survenus chez 76,9 % des 108 patients traités par céritinib à la dose recommandée de 450 mg prise avec de la nourriture dans le cadre d'une étude d'optimisation de la dose et étaient majoritairement des évènements de grade 1 (52,8 %) et de grade 2 (22,2 %). Deux patients (1,9 %) ont présenté chacun un événement de grade 3 (diarrhée et vomissements, respectivement). Une interruption du traitement à l'étude a été nécessaire pour neuf patients (8,3 %) à cause de diarrhées, de nausées ou de vomissements. Une adaptation de la dose a été nécessaire en raison de vomissements chez un patient (0,9 %). Dans la même étude, l'incidence et la sévérité des effets indésirables gastro-intestinaux ont été plus élevées chez les patients traités par 750 mg de céritinib pris à jeun (diarrhées 80,0 %, nausées 60 %, vomissements 65,5 % ; 17,3 % ont rapporté un événement de grade 3) par rapport à ceux traités à une dose de 450 mg prise avec de la nourriture (diarrhées 59,3 %, nausées 42,6 %, vomissements 38,0 % ; 1,9 % ont rapporté un événement de grade 3).
Dans les bras à 450 mg pris avec de la nourriture et 750 mg pris à jeun de cette étude d'optimisation de dose, aucun arrêt du traitement par céritinib n'a été nécessaire en raison de diarrhées, de nausées ou de vomissements (voir rubrique Effets indésirables).
Les patients doivent faire l'objet d'une surveillance et d'une prise en charge avec des traitements standards, tels que les antidiarrhéiques, les antiémétiques et une hydratation adéquate, si cliniquement indiqué. Si nécessaire, le traitement doit être interrompu ou la posologie réduite (voir rubriques Posologie et mode d'administration et Effets indésirables). Si des vomissements surviennent au cours du traitement, le patient ne doit pas prendre de dose supplémentaire, mais il doit poursuivre avec la prochaine dose programmée.
Hyperglycémie
Des cas d'hyperglycémie (tous grades confondus) ont été rapportés chez moins de 10 % des patients traités par céritinib dans le cadre des études cliniques ; une hyperglycémie de grade 3-4 a été rapportée chez 5,4 % des patients. Le risque d'hyperglycémie était plus élevé chez les patients diabétiques et/ou sous corticothérapie.
La glycémie à jeun doit être mesurée avant l'instauration du traitement par céritinib puis de manière périodique lorsque cela est cliniquement justifié. Un traitement antihyperglycémique doit être instauré ou optimisé si cliniquement indiqué (voir rubriques Posologie et mode d'administration et Effets indésirables).
Augmentation de la lipasémie et/ou de l'amylasémie
Des augmentations de la lipasémie et/ou de l'amylasémie ont été observées chez des patients traités par céritinib dans des essais cliniques. Les patients doivent être surveillés et un dosage de la lipasémie et de l'amylasémie doit être réalisé avant le début du traitement par céritinib puis, par la suite, périodiquement si cela est justifié cliniquement (voir rubriques Posologie et mode d'administration et Effets indésirables). Des cas de pancréatite ont été rapportés chez des patients traités par céritinib (voir rubrique Effets indésirables).
Teneur en sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par gélule, c.-à-d. qu'il est essentiellement « sans sodium ».
Résumé du profil de tolérance
Les effets indésirables décrits ci-dessous reflètent l'exposition à 750 mg de céritinib une fois par jour à jeun, de 925 patients atteints d'un CBNPC avancé avec réarrangement du gène anaplastic lymphoma kinase (ALK-positif) lors de sept études cliniques dont deux études de phase 3, randomisées, contre traitement actif (études A2301 et A2303).
La durée médiane d'exposition à 750 mg de céritinib à jeun a été de 44,9 semaines (intervalle : 0,1 à 200,1 semaines).
Les effets indésirables dont l'incidence a été supérieure ou égale à 10% chez les patients traités par 750 mg de céritinib à jeun étaient : diarrhées, nausées, vomissements, fatigue, anomalies du bilan hépatique, douleurs abdominales, diminution de l'appétit, perte de poids, constipation, augmentation de la créatininémie, rash, anémie et troubles œsophagiens.
Les effets indésirables de grade 3 ou 4 dont l'incidence a été supérieure ou égale à 5 % chez les patients traités par 750 mg de céritinib à jeun étaient : anomalies du bilan hépatique, fatigue, vomissements, hyperglycémie, nausées et diarrhées.
Dans l'étude d'optimisation de la dose A2112 (ASCEND-8) chez les patients préalablement traités et non-traités atteints d'un CBNPC avancé ALK-positif, le profil de tolérance global de céritinib à la dose recommandée de 450 mg avec de la nourriture (N=108) était cohérent avec celui de céritinib à la dose de 750 mg à jeun (N=110), excepté pour les effets indésirables gastro-intestinaux qui ont été réduits, tout en atteignant, à l'état d'équilibre, une exposition comparable (voir rubrique Propriétés pharmacodynamiques et la sous- rubrique « Effets indésirables gastro-intestinaux » ci-dessous).
Tableau des effets indésirables
Le tableau 2 présente la catégorie de fréquence des effets indésirables rapportés avec céritinib chez des patients traités à la dose de 750 mg à jeun (N=925) au cours de sept études cliniques. La fréquence des EI gastro-intestinaux sélectionnés (diarrhées, nausées et vomissements) est établie sur la population des patients traités avec une dose de 450 mg prise une fois par jour avec de la nourriture (N=108).
Les effets indésirables sont répertoriés par classe de systèmes d'organes selon la classification MedDRA. Au sein de chaque classe de systèmes d'organes, les effets indésirables sont classés par ordre décroissant de fréquence. De plus, la catégorie de fréquence correspondant à chaque effet indésirable repose sur la convention suivante (CIOMS III) : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque catégorie de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 2 Effets indésirables observés chez les patients traités par céritinib
| Classe de systèmes d'organes | Céritinib N = 925 % | Catégorie de fréquence |
| Affections hématologiques et du système lymphatique | ||
| Anémie | 15,2 | Très fréquent |
| Troubles du métabolisme et de la nutrition | ||
| Diminution de l'appétit | 39,5 | Très fréquent |
| Hyperglycémie | 9,4 | Fréquent |
| Hypophosphatémie | 5,3 | Fréquent |
| Affections oculaires | ||
| Troubles de la visiona | 7,0 | Fréquent |
| Affections cardiaques | ||
| Péricarditeb | 5,8 | Fréquent |
| Bradycardiec | 2,3 | Fréquent |
| Affections respiratoires, thoraciques et médiastinales | ||
| Pneumopathied | 2,1 | Fréquent |
| Affections gastro-intestinales | ||
| Diarrhéee | 59,3 | Très fréquent |
| Nauséese | 42,6 | Très fréquent |
| Vomissementse | 38,0 | Très fréquent |
| Douleurs abdominalesf | 46,1 | Très fréquent |
| Constipation | 24,0 | Très fréquent |
| Troubles œsophagiensg | 14,1 | Très fréquent |
| Pancréatites | 0,5 | Peu fréquent |
| Affections hépatobiliaires | ||
| Tests de fonction hépatique anormauxh | 2,2 | Fréquent |
| Hépatotoxicitéi | 1,1 | Fréquent |
| Affections de la peau et du tissu sous-cutané | ||
| Rashj | 19,6 | Très fréquent |
| Affections du rein et des voies urinaires | ||
| Insuffisance rénalek | 1,8 | Fréquent |
| Altération de la fonction rénalel | 1,0 | Fréquent |
| Troubles généraux et anomalies au site d'administration | ||
| Fatiguem | 48,4 | Très fréquent |
| Investigations | ||
| Anomalies du bilan hépatiquen | 60,5 | Très fréquent |
| Perte de poids | 27,6 | Très fréquent |
| Augmentation de la créatininémie | 22,1 | Très fréquent |
| Intervalle QT prolongé à l'électrocardiogramme | 9,7 | Fréquent |
| Lipase augmentée | 4,8 | Fréquent |
| Amylase augmentée | 7,0 | Fréquent |
| Les termes généraux ci-dessous englobent les cas rapportés suivants : a Troubles de la vision (déficience visuelle, vision trouble, photopsie, corps flottants du vitré, baisse de l'acuité visuelle, trouble de l'accommodation, presbytie) b Péricardite (épanchement péricardique, péricardite) c Bradycardie (bradycardie, bradycardie sinusale) d Pneumopathie (pneumopathie interstitielle diffuse, pneumopathie) e La fréquence de ces EI gastro-intestinaux sélectionnés (diarrhées, nausées et vomissements) est basée sur la population des patients traités avec la dose recommandée de 450 mg de céritinib prise avec de la nourriture (N=108) dans l'étude A2112 (ASCEND-8) (voir paragraphe ci-dessous « Effets indésirables gastro-intestinaux ») f Douleurs abdominales (douleur abdominale, douleur abdominale haute, gêne abdominale, gêne épigastrique) g Troubles œsophagiens (dyspepsie, reflux gastro-œsophagien, dysphagie) h Tests de fonction hépatique anormaux (fonction hépatique anormale, hyperbilirubinémie) i Hépatotoxicité (lésion hépatique d'origine médicamenteuse, hépatite cholestatique, lésion hépatocellulaire, hépatotoxicité) j Rash (rash, dermatite acnéiforme, rash maculopapulaire) k Insuffisance rénale (insuffisance rénale aiguë, insuffisance rénale) l Altération de la fonction rénale (azotémie, atteinte de la fonction rénale) m Fatigue (fatigue, asthénie) n Anomalies du bilan hépatique (augmentation du taux d'alanine aminotransférase, augmentation du taux d'aspartate aminotransférase, augmentation du taux de gamma-glutamyl transférase, augmentation de la bilirubinémie, augmentation du taux des transaminases, augmentation du taux d'enzymes hépatiques, test de la fonction hépatique anormal, test de la fonction hépatique augmenté, augmentation du taux de phosphatases alcalines sanguines) | ||
Patients âgés (≥ 65 ans)
Dans l'ensemble des sept études cliniques, 168 des 925 patients (18,2 %) traités par céritinib étaient âgés de 65 ans ou plus. Le profil de tolérance observé chez les patients âgés de 65 ans ou plus était similaire à celui observé chez les patients de moins de 65 ans (voir rubrique Posologie et mode d'administration). Il n'y a pas de données de sécurité disponible chez les patients âgés de plus de 85 ans.
Hépatotoxicité
Des élévations simultanées d'ALAT ou d'ASAT supérieures à 3 x LSN et de la bilirubine totale supérieure à 2 x LSN sans élévation de phosphatases alcalines ont été observées chez moins de 1 % des patients traités par céritinib dans les études cliniques. 25% des patients ont présenté des élévations d'ALAT de grade 3 ou 4. Les atteintes hépatiques ont été contrôlées par une interruption du traitement ou une diminution de la posologie chez 40,6 % des patients. Un arrêt définitif a été nécessaire chez1 % des patients dans les études cliniques avec céritinib (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Des bilans de la fonction hépatique incluant des dosages d'ALAT, d'ASAT et de la bilirubine totale doivent être réalisés avant le début du traitement par céritinib, toutes les 2 semaines durant les trois premiers mois de traitement, puis mensuellement, avec un suivi plus fréquent lors d'élévations de grade 2, 3, ou 4. Les anomalies des bilans hépatiques doivent être suivies et prises en charge selon les recommandations décrites en rubrique Posologie et mode d'administration et 4.4.
Effets indésirables gastro-intestinaux
Les nausées, diarrhées et vomissements ont été parmi les effets gastro-intestinaux les plus fréquemment rapportés. Dans l'étude d'optimisation de la dose A2112 (ASCEND-8) chez les patients préalablement traités et non traités atteints d'un CBNPC avancé ALK-positif, à la dose recommandée de 450 mg de céritinib prise avec de la nourriture (N=108), les effets indésirables diarrhées, nausées et vomissements étaient principalement de grade 1 (52,8 %) et de grade 2 (22,2 %). Un épisode de diarrhée et de vomissement de grade 3 ont été rapportés chacun chez deux patients différents (1,9 %). Les effets gastro-intestinaux ont été contrôlés principalement par la prise de traitements concomitants notamment des antiémétiques/anti-diarrhéiques. Une interruption du traitement à l'étude a été nécessaire pour neuf patients (8,3 %) à cause de diarrhées, de nausées ou de vomissements. Un patient (0,9 %) a nécessité une adaptation de dose. Dans les bras 450 mg avec de la nourriture et 750 mg à jeun, aucun patient n'a eu de diarrhées, de nausées ou de vomissements nécessitant un arrêt du traitement à l'étude. Dans la même étude, l'incidence et la sévérité des effets indésirables gastro- intestinaux ont été réduites chez les patients traités par 450 mg de céritinib pris avec de la nourriture (diarrhées 59,3 %, nausées 42,6 %, vomissements 38 % ; 1,9 % ont rapporté un événement de grade 3) par rapport à ceux traités à une dose de 750 mg à jeun (diarrhées 80,0 %, nausées 60,0 %, vomissements 65,5 % ; 17,3 % ont rapporté un événement de grade 3). Les patients doivent être pris en charge selon les recommandations décrites en rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi.
Allongement de l'intervalle QT
Un allongement de l'intervalle QTc a été observé chez certains patients traités par céritinib. Au cours des sept études cliniques, 9,7 % des patients traités par céritinib ont présentés des allongements de l'intervalle QT (de tous grades) incluant des grades 3 ou 4 chez 2,1 % des patients. Ces effets ont nécessité une diminution de la posologie ou une interruption du traitement chez 2,1 % des patients et ont entraîné un arrêt du traitement chez 0,2 %.
Le traitement par céritinib n'est pas recommandé chez les patients présentant un syndrome du QT long congénital ou prenant des médicaments connus pour allonger l'intervalle QTc (voir rubriques Mises en garde spéciales et précautions d'emploi et Interactions avec d'autres médicaments et autres formes d'interactions). Une attention particulière est nécessaire lorsque le céritinib est administré à des patients présentant un risque accru de présenter une torsade de pointe durant un traitement avec un médicament allongeant l'intervalle QT.
La survenue d'allongement de l'intervalle QT doit être surveillée et les patients doivent être pris en charge selon les recommandations décrites en rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi.
Bradycardie
Au cours des sept études cliniques, des bradycardies et/ou des bradycardies sinusales (fréquence cardiaque inférieure à 60 bpm), (toutes de grade 1) ont été rapportées chez 2,3 % des patients. Ces effets ont nécessité une diminution de la posologie ou une interruption chez 0,2 % des patients. Aucun de ces effets n'a entraîné d'arrêt du traitement par céritinib. L'utilisation concomitante de médicaments bradycardisant doit être évaluée avec attention. Les patients développant une bradycardie symptomatique doivent être pris en charge selon les recommandations décrites en rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi.
Pneumopathie interstitielle diffuse/Pneumopathie
Des pneumopathies interstitielles diffuses (PID)/pneumopathies sévères, menaçant le pronostic vital ou d'évolution fatale ont été observées chez des patients traités par céritinib. Au cours des sept études cliniques, des PID/pneumopathies de tous grades ont été rapportées chez 2,1 % des patients traités par céritinib et 1,2% des patients ont développé des grades 3 ou 4. Ces effets ont conduit à une diminution de posologie ou à une interruption de traitement chez 1,1% des patients et à l'arrêt du traitement chez 0,9 % des patients. Les patients présentant des symptômes suggérant une PID/pneumopathie doivent être surveillés. Les causes alternatives de PID/pneumopathie doivent être exclues (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Hyperglycémie
Des hyperglycémies (de tous grades) ont été rapportées chez 9,4 % des patients traités par céritinib au cours des sept études cliniques ; 5,4% des patients ont présenté des grades 3 ou 4. Une diminution de la posologie ou une interruption du traitement ont été nécessaires chez 1,4 % des patients, et un arrêt du traitement chez 0,1 % des patients. Le risque d'hyperglycémie était plus important chez les patients présentant un diabète sucré et/ou lors d'une utilisation concomitante de corticoïdes. Un suivi de la glycémie à jeun est requis avant le début du traitement par céritinib puis régulièrement si cliniquement indiqué. Un traitement anti-hyperglycémiant doit être instauré ou optimisé lorsque cela est justifié (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
SURVEILLANCE du traitement :
- Hépatique : un bilan hépatique (dosage des ALAT, ASAT et de la bilirubine totale) doit être réalisé avant l'instauration du traitement, toutes les 2 semaines les 3 premiers mois, puis une fois par mois.
- Pulmonaire : tout symptôme pulmonaire évocateur d'une pneumopathie.
- Cardiaque : fréquence cardiaque et pression artérielle.
- Glycémie à jeun : mesurer avant l'instauration du traitement puis de manière périodique lorsque cela est cliniquement justifié.
- Lipasémie et amylasémie : avant le début du traitement puis, par la suite, périodiquement si cela est justifié cliniquement.
PREVENIR IMMEDIATEMENT LE MEDECIN en cas de :
-
fatigue, démangeaisons cutanées, coloration jaune de la peau ou du
blanc des yeux, nausées ou vomissements, diminution de l'appétit,
douleur du côté droit de l'abdomen, urines foncées ou brunes,
saignements ou bleus plus fréquents que la normale,
- apparition ou
aggravation de toux, avec ou sans crachats, fièvre, douleurs au niveau
du thorax, difficultés à respirer ou essoufflement,
- douleur ou
gêne dans la poitrine, modification du rythme cardiaque (rapide ou
lent), étourdissements, évanouissements, vertiges, bleuissement des
lèvres, essoufflement, gonflement des membres inférieurs ou de la peau,
- diarrhée, nausées, vomissements sévères,
- douleurs importantes de la partie haute de l'estomac,
- soif excessive ou envie d'uriner plus fréquente.
ARRETER LE TRAITEMENT et PREVENIR IMMEDIATEMENT UN MEDECIN en cas de :
- Difficultés à respirer ou à avaler.
- Gonflement du visage, des lèvres, de la langue ou de la gorge.
- Démangeaisons sévères de la peau, avec une éruption de plaques rouges ou des petits boutons.
Les femmes DOIVENT UTILISER une méthode de contraception pendant le traitement et pendant 3 mois après son arrêt.
NE PAS PRENDRE de préparations à base de plantes contenant du millepertuis (Hypericum perforatum).
NE PAS CONSOMMER de pamplemousse ou de jus de pamplemousse.
PRUDENCE en cas de conduite de véhicules ou d'utilisation de machines (troubles visuels ou sensation de fatigue).
Femmes en âge de procréer/Contraception
Les femmes en âge de procréer doivent être informées de la nécessité d'utiliser une méthode efficace de contraception pendant toute la durée du traitement par céritinib et pendant au moins 3 mois après l'arrêt du traitement (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Grossesse
Il n'existe pas de données ou il existe des données limitées sur l'utilisation du céritinib chez la femme enceinte.
Les études effectuées chez l'animal sont insuffisantes pour permettre de conclure sur la toxicité sur la reproduction (voir rubrique Données de sécurité préclinique).
Céritinib ne doit pas être utilisé pendant la grossesse à moins que la situation clinique de la femme ne justifie le traitement par céritinib.
Allaitement
On ne sait pas si le céritinib/ses métabolites sont excrétés dans le lait maternel. Un risque pour le nouveau-né/nourrisson ne peut être exclu.
Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre/de s'abstenir du traitement avec céritinib en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la femme (voir rubrique Données de sécurité préclinique).
Fertilité
Le risque potentiel de stérilité pour l'homme et la femme demeure inconnu (voir rubrique Données de sécurité préclinique).
Agents susceptibles d'augmenter les concentrations plasmatiques de céritinib
Inhibiteurs puissants du CYP3A
Chez des sujets sains, l'administration concomitante d'une dose unique de 450 mg de céritinib à jeun et de kétoconazole (200 mg deux fois par jour pendant 14 jours), un inhibiteur puissant du CYP3A et de la P-gp, a entraîné une multiplication de l'ASCinf et de la Cmax du céritinib, respectivement par 2,9 et 1,2, par rapport aux valeurs observées avec le céritinib administré seul. Les simulations suggèrent que l'ASC à l'état d'équilibre du céritinib à doses réduites après administration concomitante de kétoconazole à 200 mg deux fois par jour pendant 14 jours était similaire à l'ASC à l'état d'équilibre du céritinib seul. L'utilisation concomitante d'inhibiteurs puissants du CYP3A pendant le traitement par céritinib doit être évitée. Si l'utilisation concomitante d'inhibiteurs puissants du CYP3A ne peut être évitée (ceci inclut entre-autres le ritonavir, le saquinavir, la télithromycine,le kétoconazole, l'itraconazole, le voriconazole, le posaconazole et la néfazodone), la dose de céritinib doit être réduite d'approximativement un tiers, arrondie à la dose la plus proche d'un multiple de 150 mg. A l'arrêt du traitement par l'inhibiteur puissant du CYP3A, le traitement par céritinib doit être repris à la posologie initiale (avant l'initiation de l'inhibiteur puissant du CYP3A).
Inhibiteurs de la P-gp
D'après les données obtenues in vitro, le céritinib est un substrat de la glycoprotéine P (P-gp), un transporteur d'efflux. Si le céritinib est administré avec des médicaments inhibiteurs de la P-gp, une augmentation de la concentration de céritinib est probable. La prudence est recommandée en cas d'utilisation concomitante d'inhibiteurs de la P-gp, et l'apparition d'EI doit être étroitement surveillée.
Agents susceptibles de diminuer les concentrations plasmatiques de céritinib
Inducteurs puissants du CYP3A et de la P-gp
Chez des sujets sains, l'administration concomitante d'une dose unique de 750 mg de céritinib à jeun et de rifampicine (600 mg par jour pendant 14 jours), un inducteur puissant du CYP3A et de la P-gp, a entraîné une diminution de respectivement 70 % et 44 % de l'ASCinf et de la Cmax du céritinib, par rapport aux valeurs observées lorsque le céritinib est administré seul. L'administration concomitante de céritinib et d'inducteurs puissants du CYP3A et de la P-gp diminue les concentrations plasmatiques de céritinib. L'utilisation concomitante d'inducteurs puissants du CYP3A doit être évitée ; ceci inclus entre-autres la carbamazépine, le phénobarbital, la phénytoïne, la rifabutine, la rifampicine et le millepertuis (Hypericum perforatum). La prudence est recommandée en cas d'utilisation concomitante d'inducteurs de la P-gp.
Agents qui affectent le pH gastrique
In vitro, le céritinib montre une solubilité pH-dépendante et devient faiblement soluble quand ce dernier augmente. Les agents réduisant l'acidité (par exemple : inhibiteurs de la pompe à protons, anti- H2, antiacides) peuvent altérer la solubilité du céritinib et réduire sa biodisponibilité. L'administration concomitante chez des sujets sains à jeun d'une dose unique de 750 mg de céritinib avec un inhibiteur de la pompe à protons (ésoméprazole) à 40 mg par jour pendant 6 jours a diminué l'ASC du céritinib de 76 % et sa Cmax de 79 %. L'étude d'interaction médicamenteuse avait pour objectif d'observer l'impact des inhibiteurs de la pompe à protons sur l'exposition au céritinib dans la situation la plus défavorable, mais en pratique clinique cet impact des inhibiteurs de la pompe à protons sur l'exposition au céritinib semble être moins prononcé. Aucune étude spécifique permettant d'évaluer l'effet des agents réduisant l'acidité gastrique sur la biodisponibilité du céritinib à l'état d'équilibre n'a été conduite. La prudence est recommandée en cas d'utilisation concomitante avec des inhibiteurs de la pompe à protons étant donné que l'exposition au céritinib peut être diminuée. Il n'y a aucune donnée sur une utilisation concomitante avec des anti-H2 ou des antiacides. Cependant, le risque d'une diminution de la biodisponibilité du céritinib cliniquement significative est potentiellement plus faible lors de l'utilisation concomitante avec des anti-H2 s'ils sont administrés 10 heures avant ou 2 heures après la prise du céritinib, et avec les antiacides s'ils sont administrés 2 heures avant ou 2 heures après la prise du céritinib.
Agents dont la concentration plasmatique peut être modifiée par céritinib
Substrats du CYP3A et du CYP2C9
D'après les données obtenues in vitro, le céritinib est un inhibiteur compétitif du métabolisme d'un substrat du CYP3A, le midazolam, et d'un substrat du CYP2C9, le diclofénac. Une inhibition temps- dépendante du CYP3A a également été observée.
In vivo, le céritinib a été classifié comme un inhibiteur puissant du CYP3A4 et pourrait interagir avec des médicaments métabolisés par le CYP3A, ce qui pourrait entraîner une augmentation des concentrations sériques de l'autre médicament. L'administration concomitante d'une dose unique de midazolam (un substrat sensible du CYP3A) après 3 semaines d'administration de céritinib (750 mg par jour à jeun) chez des patients a augmenté l'ASCinf du midazolam (IC à 90%) de 5,4 fois (4,6 ; 6,3) par rapport au midazolam seul. L'administration concomitante de céritinib et de substrats principalement métabolisés par le CYP3A ou des substrats du CYP3A connus pour avoir une marge thérapeutique étroite (alfuzosine, amiodarone, cisapride, ciclosporine, dihydroergotamine, ergotamine, fentanyl, pimozide, quetiapine, quinidine, lovastatine, simvastatine, sildenafil, midazolam, triazolam, tacrolimus, alfentanil et sirolimus par exemple) doit être évitée et des traitements alternatifs moins sensibles à l'inhibition du CYP3A doivent être utilisés si cela est possible. Si cela est inévitable, une réduction de la dose des traitements concomitants des substrats du CYP3A avec une marge thérapeutique étroite doit être envisagée.
In vivo, le céritinib a été classifié comme un inhibiteur faible du CYP2C9. L'administration concomitante d'une dose unique de warfarine (un substrat du CYP2C9) après 3 semaines d'administration de céritinib (750 mg par jour à jeun) chez des patients a augmenté de 54% (36% ; 75%) l'ASCinf de la S-warfarine (IC à 90%) par rapport à la warfarine seule. L'administration concomitante de céritinib et de substrats principalement métabolisés par le CYP2C9 ou de substrats du CYP2C9 connus pour avoir une marge thérapeutique étroite (phénytoïne et warfarine par exemple) doit être évitée. Si cela est inévitable, une réduction de la dose des traitements concomitants substrats du CYP2C9 avec une marge thérapeutique étroite doit être envisagée. L'augmentation de la fréquence de la surveillance du rapport normalisé international (INR) peut être envisagée si l'administration concomitante de warfarine est inévitable.
Substrats du CYP2A6 et du CYP2E1
D'après les données obtenues in vitro, le céritinib est également un inhibiteur du CYP2A6 et du CYP2E1 à des concentrations cliniquement significatives. Par conséquent, le céritinib pourrait augmenter la concentration plasmatique des médicaments concomitants qui sont principalement métabolisés par ces enzymes. La prudence est recommandée en cas d'utilisation concomitante de substrats du CYP2A6 et du CYP2E1, et l'apparition d'EI doit être étroitement surveillée.
En dehors des enzymes CYP3A4, un risque d'induction d'autres enzymes régulées par les PXR ne peut être complètement exclu. L'efficacité des contraceptifs oraux peut être réduite en cas d'administration concomitante.
Substrats des transporteurs
D'après les données obtenues in vitro, le céritinib n'est pas un inhibiteur du transporteur d'efflux apical MRP2, des transporteurs d'influx hépatiques OATP1B1 et OATP1B3, des transporteurs rénaux d'anions organiques OAT1 et OAT3 ni des transporteurs rénaux de cations organiques OCT1 et OCT2 à des concentrations cliniquement significatives. Il est donc peu probable que des interactions médicamenteuses dues à l'inhibition des substrats de ces transporteurs par le céritinib surviennent. Des données in vitro font supposer que le céritinib inhibe la P-gp et la BCRP intestinales aux concentrations cliniquement pertinentes. Par conséquent, le céritinib pourrait avoir la capacité d'augmenter les concentrations plasmatiques des traitements concomitants qui sont transportés par ces protéines. La prudence est requise lors de l'utilisation concomitante de substrats de la BCRP (par exemple la rosuvastatine, le topotécan, la sulfasalazine) et de la P-gp (la digoxine, le dabigatran, la colchicine, la pravastatine) et l'apparition d'effets indésirables doit être étroitement surveillée.
Interactions pharmacodynamiques
Lors des études cliniques, un allongement de l'intervalle QT a été observé avec le céritinib. Par conséquent, le céritinib doit être utilisé avec prudence chez les patients présentant ou susceptibles de présenter un allongement de l'intervalle QT, notamment les patients prenant des antiarythmiques de classe I (par exemple la quinidine, le procaïnamide, la disopyramide) ou de classe III (par exemple l'amiodarone, le sotalol, le dofétilide, l'ibutilide) ou d'autres médicaments pouvant entraîner un allongement de l'intervalle QT tels que la dompéridone, le dropéridol, la chloroquine, l'halofantrine, la clarithromycine, l'halopéridol, la méthadone, le cisapride et la moxifloxacine. Un suivi de l'intervalle QT est indiqué en association avec ces médicaments (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Interactions avec les aliments et les boissons
Céritinib doit être pris avec de la nourriture. La biodisponibilité du céritinib augmente en présence d'aliments.
Pour les patients qui présentent une affection médicale concomitante et qui sont dans l'incapacité de prendre céritinib avec de la nourriture, un schéma thérapeutique alternatif continu peut être la prise de céritinib l'estomac vide, aucune nourriture ne doit être ingérée pendant au moins deux heures avant et une heure après la prise de la dose. Les patients ne doivent pas alterner entre des prises de doses à jeun et avec de la nourriture. La posologie doit être correctement ajustée, c'est-à-dire qu'en cas de prise du traitement avec de la nourriture à une posologie de 450 mg ou 300 mg, elle doit être augmentée respectivement à 750 mg ou 450 mg en cas de prise du traitement l'estomac vide (voir rubrique Propriétés pharmacocinétiques) et à la posologie de 150 mg avec de la nourriture, le traitement doit être arrêté. Pour les ajustements de doses suivants et les recommandations pour la gestion des effets indésirables, veuillez suivre le tableau 1 (voir rubrique Posologie et mode d'administration). La dose maximum à jeun autorisée est de 750 mg (voir rubrique Propriétés pharmacocinétiques).
Les patients doivent être informés qu'ils doivent éviter la consommation de pamplemousse ou du jus de pamplemousse qui pourraient inhiber le CYP3A dans la paroi intestinale et augmenter la biodisponibilité du céritinib.
Le traitement par céritinib doit être instauré et supervisé par un médecin expérimenté dans l'utilisation des médicaments anticancéreux.
Test ALK
Une méthode d'analyse d'ALK spécifique et validée est nécessaire pour sélectionner les patients ayant un CBNPC ALK-positif (voir rubrique Propriétés pharmacodynamiques).
Le statut ALK-positif du CBNPC doit être confirmé avant l'instauration du traitement par céritinib. La recherche du statut ALK du CBNPC doit être réalisée par des laboratoires ayant des compétences reconnues dans l'utilisation de ces technologies spécifiques.
Posologie
La posologie recommandée de céritinib est de 450 mg une fois par jour, par voie orale avec de la nourriture, à la même heure chaque jour.
La dose maximale recommandée avec de la nourriture est de 450 mg une fois par jour par voie orale. Le traitement doit être poursuivi aussi longtemps qu'un bénéfice clinique est observé.
En cas d'oubli d'une dose, le patient doit rattraper la dose oubliée sauf si la dose suivante est prévue dans moins de 12 heures.
Si des vomissements surviennent au cours du traitement, le patient ne doit pas prendre une dose supplémentaire, mais doit continuer avec la prochaine dose prévue.
Céritinib doit être arrêté chez les patients ne pouvant tolérer la dose de 150 mg par jour prise avec de la nourriture.
Adaptation posologique due à des effets indésirables
Une interruption temporaire du céritinib et/ou une réduction de la posologie peuvent être nécessaires en fonction de la sécurité d'emploi et de la tolérance de chaque patient. Si une réduction de la posologie est nécessaire en raison d'un effet indésirable du médicament (EI) ne figurant pas dans le Tableau 1, cette diminution doit s'effectuer par paliers de 150 mg par jour. L'identification précoce et la prise en charge rapide des EI par les mesures thérapeutiques standards doivent être envisagées.
Chez les patients traités par 450 mg de céritinib pris avec de la nourriture, 24,1 % des patients ont eu un évènement indésirable nécessitant au moins une diminution de la dose et 55,6 % des patients ont eu un évènement indésirable nécessitant au moins une interruption de la prise du traitement. Le délai médian pour la première diminution de dose quelle qu'en soit la raison était de 9,7 semaines.
Le tableau 1 résume les recommandations de réduction de la posologie, d'interruption ou d'arrêt du traitement par céritinib pour la prise en charge de certains EI.
Tableau 1 Recommandations d'adaptation posologique de céritinib et de prise en charge des effets indésirables
| Critères | Administration de céritinib |
| Nausées, vomissements ou diarrhées graves ou intolérables malgré un traitement anti- émétique ou anti-diarrhéique optimal | Interrompre le traitement par céritinib jusqu'à amélioration, puis reprendre le traitement en réduisant la dose de 150 mg. |
| Élévation du taux d'alanine aminotransférase (ALAT) ou d'aspartate aminotransférase (ASAT) > 5 fois la limite supérieure de la normale (LSN) avec un taux de bilirubine totale concomitant ≤ 2 × LSN | Interrompre le traitement par céritinib jusqu'à un retour aux valeurs de référence des taux ALAT/ASAT ou à des valeurs ≤ 3 × LSN, puis reprendre le traitement en réduisant la dose de 150 mg. |
| Élévation du taux d'ASAT ou d'ALAT > 3 × LSN accompagnée d'une élévation du taux de bilirubine totale > 2 × LSN (en l'absence de cholestase ou d'hémolyse) | Arrêter définitivement le traitement par céritinib. |
| Pneumopathie interstitielle diffuse (PID)/pneumopathie liée au traitement quel que soit le grade | Arrêter définitivement le traitement par céritinib. |
| Intervalle QT corrigé en fonction de la fréquence cardiaque (QTc) > 500 ms sur au moins deux électrocardiogrammes (ECG) distincts | Interrompre le traitement par céritinib jusqu'au retour à la valeur de référence ou à un intervalle QTc ≤ 480 ms, contrôler et corriger si nécessaire les taux d'électrolytes puis reprendre le traitement en réduisant la dose de 150 mg. |
| Intervalle QTc > 500 ms ou allongement de l'intervalle QTc > 60 ms par rapport à la valeur de référence, avec torsades de pointes, ou tachycardie ventriculaire polymorphe ou signes/symptômes d'arythmie grave | Arrêter définitivement le traitement par céritinib. |
| Bradycardiea (symptomatique, potentiellement sévère et médicalement significative, nécessitant une intervention médicale) | Interrompre le traitement par céritinib jusqu'au retour à une bradycardie asymptomatique (grade ≤ 1) ou à une fréquence cardiaque supérieure ou égale à 60 battements par minute (bpm). Évaluer les médicaments concomitants connus pour entraîner une bradycardie, y compris les médicaments antihypertenseurs. Si un traitement concomitant favorisant la bradycardie est identifié et arrêté, ou si sa posologie est réajustée, reprendre le traitement par céritinib à la dernière dose administrée dès le retour à une bradycardie asymptomatique ou à une fréquence cardiaque supérieure ou égale à 60 bpm. Si aucun traitement concomitant favorisant la bradycardie n'est identifié, ou si le traitement concomitant favorisant la bradycardie n'est pas arrêté ou sa posologie réajustée, reprendre le traitement par céritinib en réduisant la dose de 150 mg dès le retour à une bradycardie asymptomatique ou à une fréquence cardiaque supérieure ou égale à 60 bpm. |
| Bradycardiea (conséquences menaçant le pronostic vital, nécessitant une intervention urgente) | Arrêter définitivement le traitement par céritinib si aucun médicament concomitant favorisant la bradycardie n'est identifié. Si un traitement concomitant favorisant la bradycardie est identifié et arrêté, ou si sa posologie est réajustée, reprendre le traitement par céritinib en réduisant la dose de 150 mg dès le retour à une bradycardie asymptomatique ou à une fréquence cardiaque supérieure ou égale à 60 bpm, avec des contrôles fréquentsb. |
| Hyperglycémie persistante supérieure à 250 mg/dl malgré un traitement antihyperglycémique optimal | Interrompre le traitement par céritinib jusqu'à ce que l'hyperglycémie soit contrôlée de manière adéquate puis reprendre le traitement en réduisant la dose de 150 mg. Si un contrôle adéquat de la glycémie ne peut être obtenu malgrè une prise en charge médicale optimale, arrêter définitivement le traitement par céritinib. |
| Augmentation de la lipasémie ou de l'amylasémie de grade ≥ 3 | Interrompre le traitement par céritinib jusqu'au retour de la lipasémie ou de l'amylasémie à un grade ≤ 1, puis reprendre le traitement en réduisant la dose de 150 mg. |
| a Fréquence cardiaque inférieure à 60 battements par minute (bpm) b Arrêter définitivement en cas de récidive. | |
Inhibiteurs puissants du CYP3A
L'utilisation concomitante d'inhibiteurs puissants du CYP3A doit être évitée (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). Si l'utilisation concomitante d'inhibiteurs puissants du CYP3A est inévitable, la dose de céritinib doit être réduite d'approximativement un tiers (cette dose n'a pas été évaluée cliniquement), arrondie à la dose la plus proche d'un multiple de 150 mg. La tolérance doit être étroitement surveillée chez ces patients.
Afin d'éviter un éventuel sous-dosage, en cas de traitement concomitant au long cours avec un inhibiteur puissant du CYP3A4 et en l'absence d'intolérance à la réduction de dose, la posologie pourra être ré-augmentée sous réserve d'un suivi attentif de la tolérance.
A l'arrêt du traitement par l'inhibiteur puissant du CYP3A, le traitement par céritinib sera repris à la posologie initiale (avant l'initiation de l'inhibiteur puissant du CYP3A).
Substrats du CYP3A
Lorsque le céritinib est administré concomitamment à d'autres médicaments, les recommandations relatives à l'administration concomitante avec les inhibiteurs du CYP3A4 doivent être consultées dans le Résumé des Caractéristiques du Produit (RCP) de l'autre médicament.
L'administration concomitante de céritinib et des substrats principalement métabolisés par le CYP3A ou des substrats du CYP3A connus pour avoir une marge thérapeutique étroite (alfuzosine, amiodarone, cisapride, ciclosporine, dihydroergotamine, ergotamine, fentanyl, pimozide, quetiapine, quinidine, lovastatine, simvastatine, sildenafil, midazolam, triazolam, tacrolimus, alfentanil et sirolimus, par exemple) doit être évitée et des traitements alternatifs moins sensibles à l'inhibition du CYP3A doivent être utilisés si cela est possible. Si cela est inévitable, une réduction de dose des traitements concomitants substrats du CYP3A avec une marge thérapeutique étroite doit être envisagée.
Populations particulières
Insuffisance rénale
Aucune étude pharmacocinétique spécifique n'a été conduite chez les patients présentant une insuffisance rénale. Toutefois, d'après les données disponibles, l'élimination du céritinib par voie rénale est négligeable. Par conséquent, aucune adaptation posologique n'est nécessaire chez les patients atteints d'insuffisance rénale légère à modérée. La prudence est recommandée chez les patients atteints d'insuffisance rénale sévère, en raison de l'absence d'expérience clinique dans cette population de patients (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
D'après les données disponibles, le céritinib est principalement éliminé par le foie. Des précautions particulières doivent être prises lors du traitement de patients présentant une insuffisance hépatique sévère et la posologie doit être diminuée d'environ un tiers, en arrondissant à la plus proche dose multiple de 150 mg (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacocinétiques). Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère ou modérée.
Patients âgés (≥ 65 ans)
Les données limitées relatives à la sécurité et l'efficacité du céritinib chez les patients âgés de 65 ans et plus, ne semblent pas indiquer qu'une adaptation posologique soit nécessaire dans cette population (voir rubrique Propriétés pharmacocinétiques). Il n'y a pas de données disponibles chez les patients âgés de plus de 85 ans.
Population pédiatrique
La sécurité et l'efficacité du céritinib chez les enfants et adolescents âgés de 0 à 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration
Céritinib est à usage oral. Les gélules doivent être administrées par voie orale, une fois par jour avec de la nourriture, à la même heure chaque jour. Il est important que céritinib soit pris avec de la nourriture pour atteindre l'exposition appropriée. La prise de nourriture peut aller d'un repas léger à un repas complet (voir rubrique Propriétés pharmacocinétiques). Les gélules doivent être avalées entières avec de l'eau et ne doivent pas être mâchées ou écrasées.
Pour les patients qui présentent une affection médicale concomitante et qui sont dans l'incapacité de prendre céritinib avec de la nourriture, veuillez-vous référer à la rubrique Interactions avec d'autres médicaments et autres formes d'interactions.
Durée de conservation :
3 ans.
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
Aucun cas de surdosage n'a été rapporté chez l'homme. En cas de surdosage, des mesures générales de traitement symptomatique doivent être mises en place.
Classe pharmacothérapeutique : agents antinéoplastiques, inhibiteurs de la kinase du lymphome anaplastique (ALK), code ATC : L01ED02.
Mécanisme d'action
Le céritinib est un inhibiteur oral puissant et hautement sélectif de l'ALK. Le céritinib inhibe l'autophosphorylation d'ALK, la phosphorylation induite par ALK des protéines de signalisation situées en aval et la prolifération des cellules cancéreuses ALK-dépendantes aussi bien in vitro qu'in vivo.
La translocation du gène ALK détermine l'expression de la protéine de fusion et l'altération de la voie de signalisation d'ALK qui en résultent dans le CBNPC. Dans la majorité des cas de CBNPC, EML4 est le partenaire de translocation d'ALK ; ceci engendre une protéine de fusion EML4-ALK, dans laquelle le domaine protéine kinase d'ALK a fusionné avec l'extrémité N-terminale d'EML4. Le céritinib s'est avéré efficace contre l'activité d'EML4-ALK dans une lignée cellulaire de CBNPC (H2228), entraînant une inhibition de la prolifération cellulaire in vitro et une régression tumorale dans les xénogreffes dérivées de la lignée cellulaire H2228 chez la souris et le rat.
Efficacité et sécurité cliniques
Patients atteints d'un CBNPC avancé ALK-positif non préalablement traités - étude A2301 de phase 3 randomisée (ASCEND-4)
L'efficacité et la sécurité de céritinib pour le traitement des patients atteints d'un CBNPC avancé avec réarrangement du gène anaplastic lymphoma kinase (ALK-positif), qui n'ont pas préalablement reçu de traitement systémique anti-cancéreux (dont les inhibiteurs d'ALK) à l'exception de traitement néo- adjuvant ou adjuvant, ont été démontrées dans l'étude de phase 3 A2301, randomisée, en ouvert, internationale et multicentrique.
Au total, 376 patients ont été randomisés selon un rapport 1:1 (stratification en fonction de l'indice de performance OMS, d'une chimiothérapie adjuvante/néoadjuvante antérieure et la présence ou l'absence de métastases cérébrales au moment de la sélection) pour recevoir soit le céritinib (750 mg par jour, à jeun) soit une chimiothérapie (choix à la discrétion de l'investigateur - pemetrexed[500 mg/m 2] plus cisplatine [75 mg/m2] ou carboplatine [AUC 5-6] administré tous les 21 jours).
Les patients qui avaient effectué 4 cycles de chimiothérapie (phase d'induction) sans progression de la maladie avaient par la suite reçu du pemetrexed (500 mg/m2) en monothérapie de maintenance tous les 21 jours. Cent quatre-vingt-neuf (189) patients ont été randomisés dans le bras du céritinib et cent quatre-vingt-sept (187) dans le bras de la chimiothérapie.
L'âge médian était de 54 ans (intervalle : de 22 à 81 ans) ; 78,5 % des patients avaient moins de 65 ans. Un total de 57,4 % des patients étaient des femmes. 53,7 % de la population de l'étude était de type caucasien, 42,0 % asiatique, 1,6 % africain et 2,6 % d'autres origines ethniques. La majorité des patients avaient un adénocarcinome (96,5 %) et n'avaient soit jamais fumé ou étaient d'anciens fumeurs (92,0%). Le statut de performance Eastern Cooperative Oncology group (ECOG) était de 0/1/2 pour respectivement 37,0%/56,4%/6,4% des patients, et 32,2% avaient des métastases cérébrales à l'inclusion. 59,5% des patients avec des métastases cérébrales à l'inclusion n'avaient pas eu de radiothérapie cérébrale. Les patients avec des métastases du SNC (système nerveux central) symptomatiques qui étaient neurologiquement instables ou qui nécessitaient une augmentation des doses de stéroïdes au cours des 2 semaines précédant la sélection, ont été exclus de l'étude.
Les patients étaient autorisés à continuer le traitement qui leur avait été attribué dans l'étude en cas de progression, si l'investigateur estimait qu'il y avait un bénéfice clinique continu. Les patients randomisés dans le bras de la chimiothérapie pouvaient passer dans l'autre bras pour recevoir le céritinib en cas de progression de la maladie selon les critères RECIST confirmée par le Comité de Revue Indépendant à l'Aveugle (CRIA). Cent cinq (105) des 145 patients (72,4%) qui ont arrêté le traitement dans le bras de la chimiothérapie ont par la suite reçu un inhibiteur d'ALK comme premier traitement antinéoplasique. Parmi ces patients, 81 ont reçu du céritinib.
La durée de suivi médiane était de 19,7 mois (de la randomisation jusqu'à la date de cut-off).
L'étude a atteint son critère principal démontrant une amélioration statistiquement significative de la survie sans progression (SSP) évaluée par le CRIA (voir tableau 3 et figure 1). Le bénéfice obtenu en terme de SSP dans le bras du céritinib était similaire lorsqu'elle était évaluée par les investigateurs ainsi que dans les divers sous-groupes définis en fonction de l'âge, du genre, de l'origine ethnique, du statut tabagique, du statut de performance ECOG et du fardeau de la maladie.
Les données de survie globale (SG) n'étaient pas matures avec la survenue de 107 décès représentant approximativement 42,3% des événements nécessaires pour l'analyse finale de la SG.
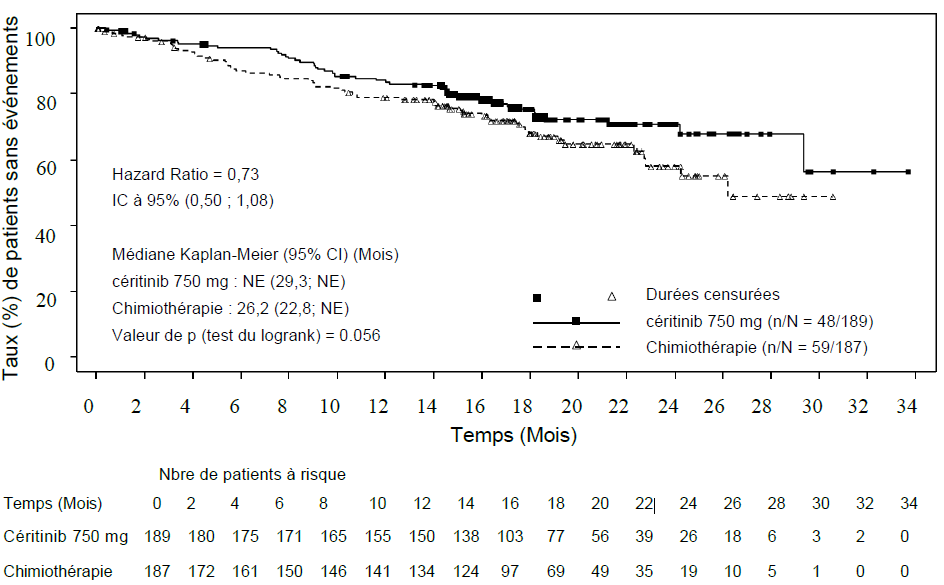
Les données d'efficacité de l'étude A2301 sont résumées dans le tableau 3, et les courbes de Kaplan- Meier pour la SSP et la SG sont présentées dans les figures 1 et 2, respectivement.
Tableau 3 ASCEND-4 (Etude A2301) - Résultats d'efficacité chez les patients avec un CBNPC avancé ALK-positif non préalablement traités
| | Céritinib (N=189) | Chimiothérapie (N=187) |
| Survie sans progression (selon le CRIA) | | |
| Nombre d'évènements, n (%) | 89 (47,1) | 113 (60,4) |
| Médiane, moisd (IC à 95%) | 16,6 (12,6 ; 27,2) | 8;1 (5,8 ; 11,1) |
| HR (IC à 95%)a | 0,55 (0,42 ; 0,73) | |
| Valeur de pb | <0,001 | |
| Survie globalec | | |
| Nombre d'évènements, n (%) | 48 (25,4) | 59 (31,6) |
| Médiane, moisd (IC à 95%) | NE (29,3 ; NE) | 26,2 (22,8 ; NE) |
| Taux de SG à 24 moisd; % (IC à 95%) | 70,6 (62,2 ; 77,5) | 58,2 (47,6 ; 67,5) |
| HR (IC à 95%)a | 0,73 (0,50 ; 1,08) | |
| Valeur de pb | 0,056 | |
| Réponse tumorale (selon le CRIA) | | |
| Taux de réponse objective (IC à 95%) | 72,5% (65,5 ; 78;7) | 26,7% (20,5 ; 33,7) |
| Durée de la réponse (selon le CRIA) | | |
| Nombre de patients répondeurs | 137 | 50 |
| Médiane, moisd (IC à 95%) | 23,9 (16,6 ; NE) | 11,1 (7,8 ; 16,4) |
| Taux de patients sans évènements à 18 moisd; % (IC à 95%) | 59,0 (49,3 ; 67,4) | 30,4 (14,1 ; 48,6) |
| HR=Hazard ratio ; IC=intervalle de confiance ; CRIA=Comité de revue indépendant en aveugle ; NE=non estimable a Sur la base d'une analyse stratifiée d'un modèle de risques proportionnels de Cox b Sur la base d'un test de log-rank stratifié c L'analyse de l'OS n'a pas été ajustée par les effets du cross-over, d Estimé en utilisant la méthode de Kaplan-Meier, | ||
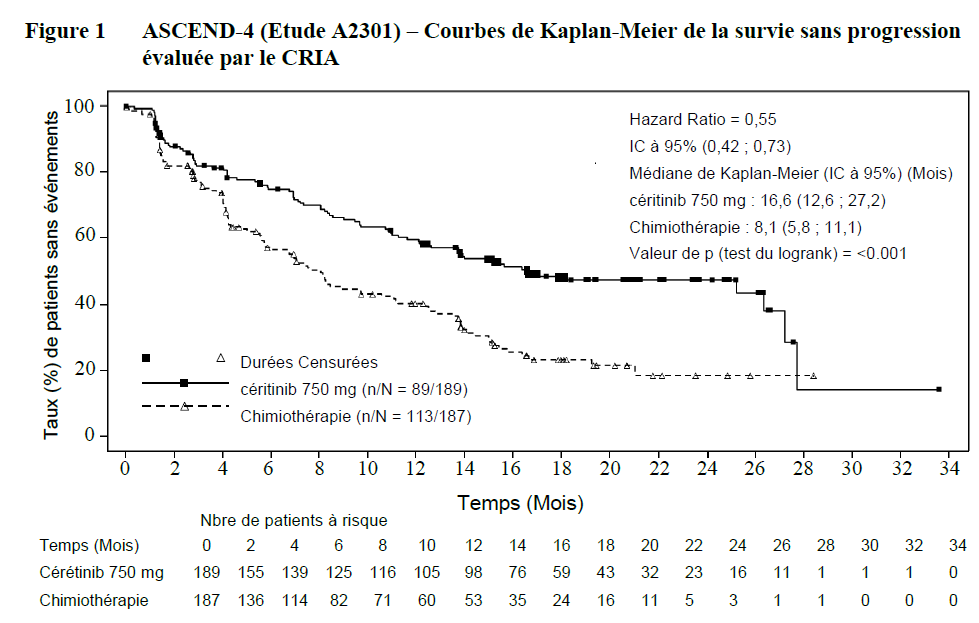
Figure 2 ASCEND-4 (Etude A2301)- Courbe de Kaplan-Meier de la survie globale en fonction du bras de traitement,
Les questionnaires de qualité de vie notés par les patients (Lung cancer symptom scale [LCSS], EORTC-QLQ-C30 [C30], EORTC QLQ-LC13 [LC13] et EQ-5D-5L) ont été complétés par au moins 80% des patients dans les bras du céritinib et de la chimiothérapie, pour tous les questionnaires et pour la plupart du temps aux moments prévus de l'étude.
Le céritinib a significativement prolongé le temps jusqu'à détérioration des symptômes d'intérêt spécifiques du cancer du poumon prédéfinis c'est-à-dire la toux, la douleur et la dyspnée (critère composite de la LCSS : HR=0,61, IC à 95% : 0,41 ;0,90, temps jusqu'à la détérioration médian NE [IC à 95% : 20,9 ; NE] dans le bras du céritinib contre 18,4 mois [13,9, NE] dans le bras de la chimiothérapie ; LC13 : HR=0,48, IC à 95% : 0,34 ; 0,69, temps jusqu'à la détérioration médian de 23,6 mois [IC à 95% : 20,7 ;NE] dans bras du céritinib contre 12,6 mois [IC à 95% : 8,9 ; 14,9] dans le bras de la chimiothérapie).
Les patients recevant le céritinib ont montré des améliorations significatives par rapport à la chimiothérapie dans les mesures de Qualité de Vie générale et de l'état de santé (LCSS [p<0,001], QLQ-C30, [p<0,001] et index EQ-5D-5L [p<0,001]).
Dans l'étude A2301, la réponse intracrânienne de 44 patients (22 patients dans le bras céritinib et 22 patients dans le bras chimiothérapie) dont les métastases cérébrales étaient mesurables à l'inclusion dans l'étude et évaluées par imagerie au moins fois après le début de l'étude, a été évaluée par le CRIA de neuroradiologie selon les critères RECIST 1.1 modifiés (c'est-à-dire jusqu'à 5 lésions cérébrales). Le taux de réponse objective intracrânien (TROI) était plus élevé dans le bras du céritinib (72,7%, IC à 95% : 49,8 ; 89,3) en comparaison au bras de la chimiothérapie (27,3%, IC à 95% : 10,7 ; 50,2).
La SSP médiane évaluée par le CRIA selon les critères RECIST 1.1 était plus longue dans le bras du céritinib par rapport au bras de la chimiothérapie pour les deux sous-groupes de patients avec métastases cérébrales et sans métastases cérébrales. La SSP médiane chez les patients avec des métastases cérébrales était de 10,7 mois (IC à 95% : 8,1 ; 16,4) contre 6,7 mois (IC à 95% : 4,1 ; 10,6) dans le bras du céritinib et de la chimiothérapie respectivement, avec un HR=0,70 (IC à 95% : 0,44 ; 1,12). La SSP médiane chez les patients sans métastases cérébrales était de 26,3 mois (IC à 95% : 15,4 ; 27,7) contre 8,3 mois (IC à 95% : 6,0 ; 13,7) dans le bras du céritinib et de la chimiothérapie respectivement, avec un HR=0,48 (IC à 95% : 0,33 ; 0,69).
Patients atteints d'un CBNPC avancé ALK-positif préalablement traités - Etude de phase 3, randomisée, A2303 (ASCEND-5)
L'efficacité et la sécurité de céritinib dans le traitement de patients atteints d'un CBNPC avancé ALK- positif ayant préalablement reçu un traitement par crizotinib, ont été démontrés dans l'étude A2303 de phase 3, en ouvert, randomisée, internationale et multicentrique.
Un total de 231 patients atteints d'un CBNPC avancé ALK-positif ayant préalablement reçu un traitement par crizotinib et par chimiothérapie (un ou deux lignes de traitement incluant un doublet de chimiothérapie base de platine) ont été inclus dans l'analyse. Cent quinze (115) patients ont été randomisés dans le bras de céritinib et cent seize (116) ont été randomisés dans le bras de la chimiothérapie (recevant soit du pemetrexed soit du docétaxel). Soixante-treize (73) patients avaient reçu du docétaxel et 40 avaient reçu du pemetrexed. Dans le bras du céritinib, 115 patients ont été traités par 750 mg une fois par jour à jeun. L'âge médian était de 54,0 ans (intervalle : de 28 à 84 ans) ; 77,1% des patients avaient moins de 65 ans. Un total de 55,8% des patients étaient des femmes. 64,5% de la population de l'étude étaient de type caucasien, 29,4% asiatique, 0,4% africain et 2,6% d'autres origines ethniques. La majorité des patients avaient un adénocarcinome (97,0%) et n'avaient soit jamais fumé ou étaient d'anciens fumeurs (96,1%). Le statut de performance ECOG était 0/1/2 chez respectivement 46,3%/47,6%/6,1% des patients, et 58,0% des patients étaient atteints de métastases cérébrales à l'inclusion. Tous les patients avaient été préalablement traités par le crizotinib. Tous les patients, excepté un, avaient également été traités préalablement une chimiothérapie (y compris un doublet à base de platine) et 11,3% des patients dans le bras du céritinib et 12,1% des patients dans le bras de la chimiothérapie avaient été préalablement traités par deux cycles de chimiothérapie pour traiter leur cancer avancé.
Les patients étaient autorisés à continuer le traitement qui leur avait été attribué dans l'étude en cas de progression si l'investigateur estimait qu'il y avait un bénéfice clinique continu. Les patients randomisés dans le bras de la chimiothérapie pouvaient passer dans l'autre bras pour recevoir céritinib en cas de progression de la maladie selon les critères RECIST et confirmée par le CRIA.
La durée de suivi médiane était de 16,5 mois (de la randomisation jusqu'à la date du cut-off).
L'étude a atteint son critère principal en démontrant une amélioration statistiquement significative de la SSP évaluée par le CRIA avec une réduction du risque estimée à 51% dans le bras du céritinib par rapport au bras de la chimiothérapie (voir Tableau 4 et Figure 3). Le bénéfice obtenu en terme de SSP dans le bras de céritinib était similaire dans les divers sous-groupes définis en fonction de l'âge, du genre, de l'ethnie, du statut tabagique, du statut de performance ECOG, et de la présence de métastases cérébrales ou de la réponse préalable au crizotinib. Le bénéfice en terme de SSP était par ailleurs confirmé par l'évaluation de l'investigateur local, et par l'analyse du taux de réponse objective (TRO) et du taux de contrôle de la maladie (TCM).
Les données de la SG étaient immatures avec 48 évènements (41,7%) dans le bras céritinib et 50 évènements (43,1%) dans le bras de la chimiothérapie, correspondant approximativement à 50% des évènements nécessaires pour l'analyse finale de la SG. De plus, 81 patients (69,8%) du bras de la chimiothérapie ont par la suite reçu céritinib comme premier traitement antinéoplasique après l'arrêt du traitement de l'étude.
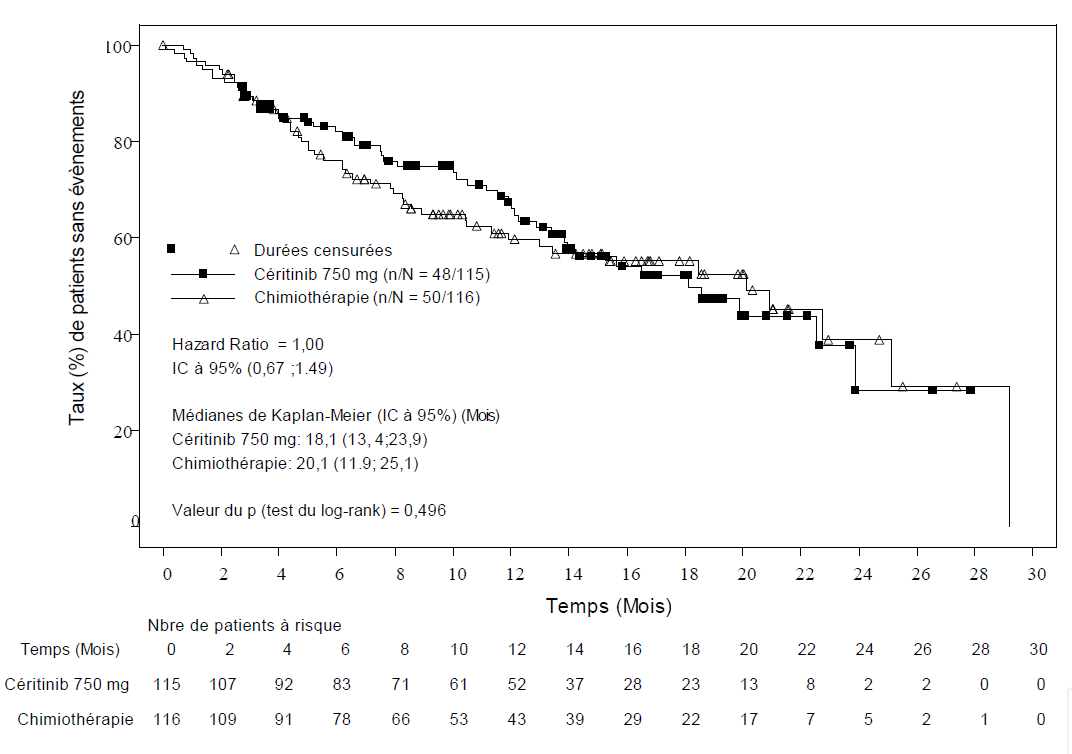
Les données d'efficacité de l'étude A2303 sont résumées dans le Tableau 4, et les courbes de Kaplan- Meier pour la SSP et la SG sont représentées respectivement dans les Figures 3 et 4.
Tableau 4 ASCEND-5 (Etude A2303) - Résultats d'efficacité chez les patients présentant un CBNPC métastatique ou avancé ALK-positif préalablement traité
| | Céritinib (N=115) | Chimiothérapie (N=116) |
| Durée de suivi Médiane (mois) (min - max) | 16,5 (2,8 - 30,9) | |
| Survie sans progression (selon le CRIA) | | |
| Nombre d'évènements; n (%) | 83 (72,2%) | 89 (76,7%) |
| Médiane; mois (IC à 95%) | 5,4 (4,1 ; 6,9) | 1,6 (1,4 ; 2,8) |
| HR (IC à 95%)a | 0,49 (0,36; 0,67) | |
| valeur de pb | <0,001 | |
| Survie globalec | | |
| Nombre d'évènements; n (%) | 48 (41,7%) | 50 (43,1%) |
| Médiane; mois (IC à 95%) | 18,1 (13,4 ; 23,9) | 20,1 (11,9 ; 25,1) |
| HR (IC à 95%)a | 1,00 (0,67 ; 1,49) | |
| valeur de pb | 0,496 | |
| Réponse tumorale (selon le CRIA) | | |
| Taux de réponse objective (IC à 95%) | 39,1% (30,2 ; 48,7) | 6,9% (3,0 ; 13,1) |
| Durée de réponse | | |
| Nombre de patients répondeurs | 45 | 8 |
| Médiane; moisd (IC à 95%) | 6,9 (5,4 ; 8,9) | 8,3 (3,5 ; NE) |
| Taux estimé de patients sans évènement à 9 moisd (IC à 95%) | 31,5% (16,7% ; 47,3%) | 45,7% (6,9% ; 79,5%) |
| HR=Hazard Ratio ; IC=intervalle de confiance, CRIA=Comité de revue indépendant en aveugle; NE=non estimable a Sur la base d'une analyse stratifiée d'un modèle de risques proportionnels de Cox b Sur la base d'un test de log-rank stratifié c L'analyse de l'OS n'a pas été ajustée par les effets du cross-over d Estimé en utilisant la méthode de Kaplan-Meier | ||
Figure 3 ASCEND-5 (Etude A2303) - Courbes de Kaplan-Meier de la survie sans progression évaluée par le CRIA
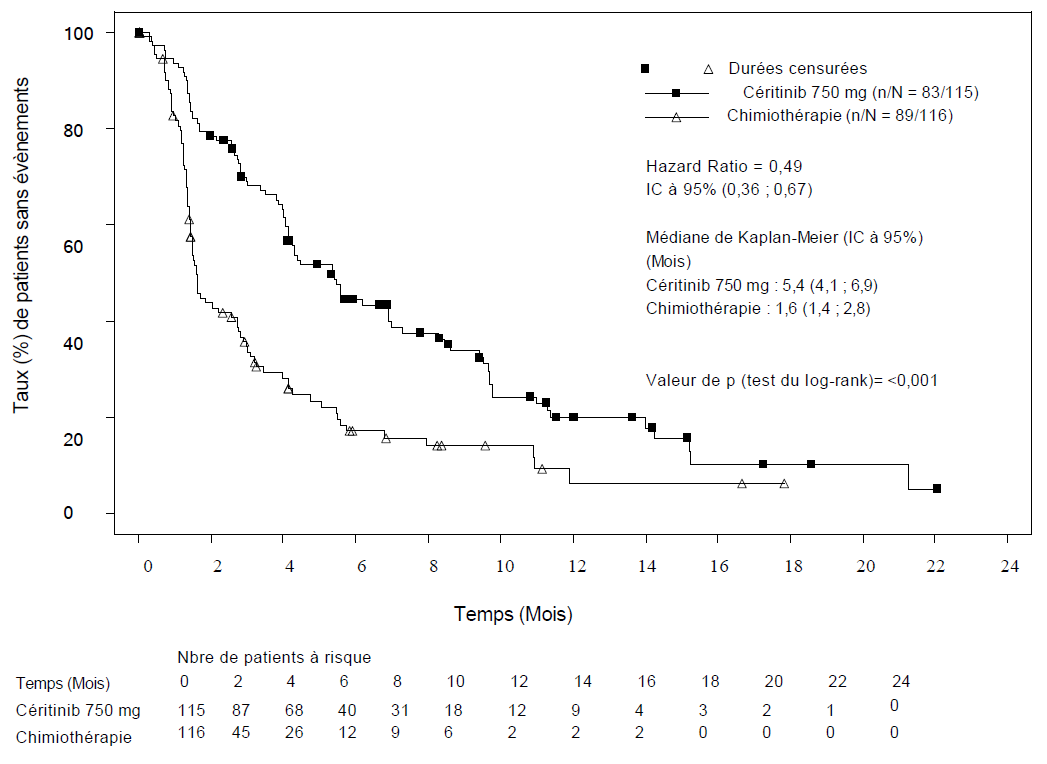
Figure 4 ASCEND-5 (Etude A2303) - Courbes de Kaplan-Meier de la survie globale du bras recevant le traitement
Dans l'étude A2303, la réponse intracrânienne a été évaluée par le CRIA de neuroradiologie selon les critères modifiés RECIST 1.1 (c'est-à-dire jusqu'à 5 lésions cérébrales) chez 133 patients présentant des métastases cérébrales à l'inclusion dans l'étude (66 patients dans le bras céritinib et 67 patients dans le bras de la chimiothérapie). Le TROI était plus élevé dans le bras du céritinib (35,3%, IC à 95% : 14,2 : 61,7) en comparaison au bras de la chimiothérapie (5,0%, IC à 95% : 0,1 ; 24,9) chez les patients avec une atteinte cérébrale mesurable et pour lesquels la maladie a été évaluée au début de l'étude et au moins fois après le début de l'étude. La SSP médiane évaluée par le CRIA selon les critères RECIST 1.1 était plus longue dans le bras du céritinib comparé au bras de la chimiothérapie dans les deux sous-groupes de patients avec et sans métastases cérébrales. La SSP médiane chez les patients avec des métastases cérébrales était de 4,4 mois (IC à 95% : 3,4 ; 6,2) contre 1,5 mois (IC à 95% : 1,3 ; 1,8) dans les bras du céritinib et de la chimiothérapie respectivement, avec un HR=0,54 (IC à 95% : 0,36 ; 0,80). La SSP médiane chez les patients sans métastases cérébrales était de 8,3 mois (IC à 95% : 4,1 ; 14,0) contre 2,8 mois (IC à 95% : 1,4 ; 4,1) dans les bras du céritinib et de la chimiothérapie respectivement, avec un HR=0,41 (IC à 95% : 0,24 ; 0,69).
Etude d'optimisation de la dose A2112 (ASCEND-8)
L'efficacité de 450 mg de céritinib pris avec de la nourriture a été évaluée lors d'une étude d'optimisation de la dose A2112 (ASCEND-8), multicentrique, ouverte. Au total, 147 patients préalablement non traités atteints d'un CBNPC ALK-positif localement avancé ou métastatique ont été randomisés pour recevoir 450 mg de céritinib une fois par jour avec de la nourriture (N=73) ou 750 mg de céritinib une fois par jour à jeun (N=74). Un principal objectif secondaire d'efficacité était le TRO évalué par le CRIA selon les critères RECIST 1.1.
Les caractéristiques de la population des patients préalablement non traités atteints d'un CBNPC ALK positif, localement avancé ou métastatique, dans les deux bras 450 mg avec prise de nourriture (N=73) et 750 mg à jeun (N=74) étaient respectivement les suivantes : un âge moyen de 54,3 et 51,3 ans, âge inférieur à 65 ans (78,1 % et 83,8 %), femmes (56,2 % et 47,3 %), caucasiens (49,3 % et 54,1 %), asiatiques (39,7 % et 35,1 %), anciens fumeurs ou n'avaient jamais fumé (90,4% et 95,9 %), indice de performance Organisation Mondiale de la Santé (OMS) à 0 ou 1 (91,7 % et 91,9 %), histologie d'adénocarcinome (98,6% et 93,2 %) et métastases cérébrales (32,9 % et 28,4 %).
Les résultats d'efficacité d'ASCEND-8 sont résumés dans le tableau 5 ci-dessous.
Tableau 5 ASCEND-8 (Etude A2112) - Résultats d'efficacité chez les patients préalablement non traités atteints de CBNPC ALK-positif localement avancé ou métastatique selon les critères de CRIA
| Paramètres d'efficacité | 450 mg de céritinib avec de la nourriture (N=73) | 750 mg de céritinib à jeun (N=74) |
| Taux de Réponse Objective (TRO: RC+RP), n (%) (IC à 95%)a | 57 (78,1) (66,9 ; 86,9) | 56 (75,7) (64,3 ; 84,9) |
| IC: Intervalle de Confiance Réponse Complète (RC), Réponse Partielle (RP) confirmée par des évaluations répétées effectuées au moins 4 semaines après l'apparition des premiers critères de réponse. Taux de réponse objective déterminé par le CRIA selon les critères RECIST 1.1 aIntervalle de confiance binomial exact de 95% | ||
Etudes à simple bras unique X2101 et A2201
L'utilisation de céritinib dans le traitement du CBNPC ALK-positif préalablement traités par un inhibiteur ALK a fait l'objet de deux études de phase 1/2, internationales, multicentriques, en ouvert et à bras unique (étude X2101 et étude A2201).
Dans l'étude X2101 un total de 246 patients atteints d'un CBNPC ALK-positif ont été traités par céritinib à une dose de 750 mg une fois par jour à jeun : 163 d'entre eux avaient préalablement été traités par un inhibiteur ALK et 83 n'avaient jamais reçu d'inhibiteur ALK. Chez les 163 patients présentant un CBNPC ALK positif préalablement traités par un inhibiteur ALK, l'âge médian était de 52 ans (intervalle : 24-80 ans) ; 86,5 % des patients avaient moins de 65 ans et les femmes représentaient 54 % des patients. La majorité des patients était de type caucasien (66 ,3 %) ou asiatique (28,8 %). 93,3 % présentait un adénocarcinome 96,9% étaient d'anciens fumeurs ou n'avait jamais fumé. Tous les patients avaient reçu antérieurement au moins une ligne de traitement avant l'inclusion dans l'étude, et 84,0 % deux lignes ou plus.
L'étude A2201 a concerné140 patients qui avaient préalablement reçu 1 à 3 lignes de chimiothérapie suivie(s) d'un traitement par crizotinib, et qui avaient progressé sous crizotinib. L'âge médian était de 51 ans (intervalle : 29-80 ans) et 87,1 % des patients avaient moins de 65 ans et les femmes représentaient 50,0 %. La majorité des patients était de type caucasien (60,0 %) ou asiatique (37,9 %). 92,1 % des patients présentait un adénocarcinome.
Le tableau 6 résume les principales données d'efficacité des deux études. Les données finales de survie globale (SG) sont présentées pour l'étude A2201. Pour l'étude X2101, les données de SG n'étaient pas encore matures au moment de l'analyse.
Tableau 6 CBNPC ALK-positif avancé - synthèse des données d'efficacité des études X2101 et A2201
| | Étude X2101 céritinib 750 mg | Étude A2201 céritinib 750 mg |
| | N = 163 | N = 140 |
| Durée du suivi Médiane (mois) (min - max) | 10,2 (0,1-24,1) | 14,1 (0,1-35,5) |
| Taux de réponse objective | | |
| Selon l'investigateur (IC à 95%) | 56,4% (48,5 ; 64,2) | 40,7% (32,5 ; 49,3) |
| Selon le CRIA (IC à 95%) | 46,0% (38,2 ; 54,0) | 35,7% (27,8 ; 44,2) |
| Durée de la réponse* | | |
| Selon l'investigateur (mois, IC à 95%) | 8,3 (6,8 ; 9,7) | 10,6 (7,4 ; 14,7) |
| Selon le CRIA (mois, IC à 95%) | 8,8 (6,0 ; 13,1) | 12,9 (9,3 ; 18,4) |
| Survie sans progression | | |
| Selon l'investigateur (mois, | 6,9 | 5,8 |
| IC à 95%) | (5,6 ; 8,7) | (5,4 ; 7,6) |
| Selon le CRIA (mois, | 7,0 | 7,4 |
| IC à 95%) | (5,7 ; 8,7) | (5,6 ; 10,9) |
| Survie globale (mois, IC à 95%) | 16.7 (14.8 ; NE) | 15,6 (13,6 ; 24,2) |
| NE = non estimable Étude X2101: réponses évaluées selon les critères RECIST 1.0 Étude A2201: réponses évaluées selon les critères RECIST 1.1 *Inclut uniquement les patients ayant présenté une RC ou RP confirmée | ||
Dans les études X2101 et A2201, 60,1 % et 71,4 % des patients, respectivement, présentaient des métastases cérébrales. Le TRO, la DR et la SSP (selon l'évaluation du CRIA) chez les patients avec des métastases à l'inclusion étaient comparables à ceux rapportés dans l'ensemble de la population de ces études.
Histologie autre qu'un adénocarcinome
Les informations disponibles sur les patients atteints d'un CBNPC ALK-positif d'histologie autre qu'un adénocarcinome sont limitées.
Patients âgés
Il y a peu de données d'efficacité chez les patients âgés. Aucune donnée d'efficacité n'est disponible chez les patients de plus de 85 ans.
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec céritinib dans tous les sous-groupes de la population pédiatrique dans le cancer du poumon (à petites cellules et non à petites cellules) (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Absorption
Les pics de concentrations plasmatiques (Cmax) de céritinib sont atteints environ 4 à 6 heures après une seule administration par voie orale. Le taux d'absorption après administration par voie orale a été estimé à au moins 25 %, d'après le pourcentage de métabolites retrouvés dans les fèces. La biodisponibilité absolue du céritinib n'a pas été déterminée.
L'exposition systémique au céritinib a été augmentée lorsque celui-ci est pris avec des aliments. Les valeurs de l'ASCinf du céritinib ont été supérieures d'environ 58 % et 73 % (et celles de la Cmax d'environ 43 % et 41 %) chez les sujets sains lorsqu'une dose unique de 500 mg de céritinib a été administrée avec un repas pauvre en lipides (contenant environ 330 kilo calories et 9 grammes de lipides) et un repas riche en graisses (contenant environ 1000 kilo calories et 58 grammes de lipides), respectivement, par rapport à une prise à jeun.
Dans l'étude d'optimisation de la dose A2112 (ASCEND-8) comparant les patients recevant 450 mg ou 600 mg de céritinib par jour avec de la nourriture (environ 100 à 500 kilo calories et 1,5 à 15 grammes de lipides) à des patients recevant 750 mg par jour à jeun (dose et conditions d'administration avec la nourriture initialement autorisées), il n'y a pas eu de différence cliniquement significative de l'exposition systémique au céritinib à l'état d'équilibre entre le bras à 450 mg avec de la nourriture (N=36) comparé au bras à 750 mg à jeun (N=31), avec seulement de faibles augmentations de l'ASC à l'état d'équilibre (IC à 90%) de 4% (-13% ; 24%) et de la Cmax (IC à 90%) de 3% (-14% ; 22%). En revanche, l'ASC à l'état d'équilibre (IC à 90%) et la Cmax (IC à 90%) pour le bras à 600 mg avec de la nourriture (N=30) ont augmenté de 24% (3% ; 49%) et de 25% (4% ; 49%), respectivement, par rapport au bras à 750 mg à jeun. La dose maximale recommandée de céritinib est de 450 mg pris par voie orale une fois par jour avec de la nourriture (voir rubrique Posologie et mode d'administration).
Après l'administration d'une dose unique de céritinib par voie orale, l'exposition plasmatique au céritinib, représentée par Cmax et ASCt, a augmenté proportionnellement à la dose sur l'intervalle de doses allant de 50 à 750 mg à jeun. En revanche, la concentration pré-dose (Cmin) après une administration quotidienne répétée semblait augmenter de façon plus que proportionnelle à la dose.
Distribution
In vitro, la liaison du céritinib aux protéines plasmatiques humaines est d'environ 97 %, quelle que soit la concentration, pour des concentrations allant de 50 ng/ml à 10 000 ng/ml. Par ailleurs, le céritinib montre une distribution légèrement plus importante dans les érythrocytes que dans le plasma, avec un rapport moyen sang/plasma in vitro de 1,35. Les études in vitro suggèrent que le céritinib est un substrat de la glycoprotéine P (P-gp), mais pas de la protéine BCRP (Breast Cancer Resistance Protein) ni de la protéine MRP2 (Multi-Resistance Protein 2). In vitro, la perméabilité passive apparente du céritinib s'est avérée faible.
Chez le rat, le céritinib traverse la barrière hémato-encéphalique intacte avec un rapport d'exposition cerveau/sang (ASCinf) d'environ 15 %. Aucune donnée n'est disponible sur le rapport d'exposition cerveau/sang chez l'homme.
Biotransformation
Les études in vitro ont montré que le CYP3A était la principale enzyme impliquée dans la clairance métabolique du céritinib.
Après l'administration par voie orale d'une dose unique de 750 mg à jeun de céritinib radioactif, le céritinib était le principal composant circulant dans le plasma humain. Au total, 11 métabolites étaient en circulation dans le plasma à de faibles concentrations, avec une contribution moyenne à l'ASC de la radioactivité ≤ 2,3 % pour chaque métabolite. Les principales voies de biotransformation identifiées chez les sujets sains étaient la mono-oxygénation, l'O-désalkylation et la N-formylation. Les voies de biotransformation secondaires impliquant les produits primaires de la biotransformation étaient la glucuronidation et la déshydrogénation. L'ajout d'un groupement thiol au céritinib O-désalkylé a également été observé.
Élimination
Après l'administration de doses uniques de céritinib par voie orale à jeun, la moyenne géométrique de la demi-vie plasmatique terminale apparente (T½) du céritinib se situait entre 31 et 41 heures sur l'intervalle de doses allant de 400 à 750 mg. Après l'administration quotidienne de céritinib par voie orale, l'état d'équilibre est atteint en 15 jours environ puis reste stable, avec une moyenne géométrique du rapport d'accumulation de 6,2 après 3 semaines d'administration quotidienne. La moyenne géométrique de la clairance apparente (CL/F) du céritinib était plus basse à l'état d'équilibre (33,2 litres/heure) après administration par voie orale d'une dose quotidienne de 750 mg qu'après administration par voie orale d'une dose unique de 750 mg (88,5 litres/heure), ce qui laisse penser que la pharmacocinétique du céritinib n'est pas linéaire dans le temps.
Le céritinib et ses métabolites sont principalement excrétés dans les fèces. En moyenne, 68 % d'une dose de céritinib administrée par voie orale se retrouvent dans les fèces sous forme inchangée.
Seulement 1,3 % de la dose administrée par voie orale se retrouve dans les urines.
Populations particulières
Insuffisance hépatique
L'effet de l'insuffisance hépatique sur la pharmacocinétique du céritinib en dose unique (750 mg à jeun) a été évalué chez des sujets avec une insuffisance hépatique légère (Classe A de Child-Pugh ; N = 8), modérée (Classe B de Child-Pugh ; N = 7) ou sévère (Classe C de Child-Pugh ; N = 7) et chez 8 sujets sains avec une fonction hépatique normale. La moyenne géométrique de l'ASCinf (ASCinf de la fraction libre) du céritinib a été augmentée de 18% (35%) et de 2% (22%) chez les sujets présentant une insuffisance hépatique légère et modérée, respectivement, par rapport aux sujets ayant une fonction hépatique normale.
La moyenne géométrique de l'ASCinf (ASCinf de la fraction libre) du céritinib a été augmentée de 66% (108%) chez les sujets avec une insuffisance hépatique sévère par rapport aux sujets ayant une fonction hépatique normale (voir rubrique Posologie et mode d'administration). Aucune étude spécifique de pharmacocinétique à l'état d'équilibre n'a été conduite chez les patients présentant une insuffisance hépatique.
Insuffisance rénale
Aucune étude pharmacocinétique spécifique n'a été conduite chez les patients présentant une insuffisance rénale. Les données disponibles suggèrent que l'élimination du céritinib par les reins est négligeable (1,3 % d'une dose unique administrée par voie orale).
Dans une analyse pharmacocinétique de population portant sur 345 patients atteints d'insuffisance rénale légère (CLcr de 60 à < 90 ml/min), 82 patients atteints d'insuffisance rénale modérée (CLcr de 30 à < 60 ml/min) et 546 patients présentant une fonction rénale normale (CLcr ≥ 90 ml/min), l'exposition au céritinib était similaire chez les patients atteints d'insuffisance rénale légère ou modérée et ceux présentant une fonction rénale normale, ce qui suggère qu'aucune adaptation posologique n'est nécessaire chez les patients atteints d'insuffisance rénale légère à modérée. Les patients atteints d'insuffisance rénale sévère (CLcr < 30 ml/min) n'ont pas été inclus dans les études cliniques de céritinib (voir rubrique Posologie et mode d'administration).
Effets de l'âge, du sexe et de l'origine ethnique
Des analyses pharmacocinétiques de population ont montré que l'âge, le sexe et l'origine ethnique n'avaient aucune influence cliniquement significative sur l'exposition au céritinib.
Électrophysiologie cardiaque
Le risque d'allongement de l'intervalle QT dû au céritinib a été évalué dans sept études cliniques menées avec céritinib. Des ECG en série ont été réalisés après l'administration d'une dose unique et à l'état d'équilibre pour évaluer l'effet du céritinib sur l'intervalle QT chez 925 patients traités par 750 mg de céritinib une fois par jour à jeun. Une analyse catégorique des valeurs aberrantes des données d'ECG a mis en évidence l'apparition d'un intervalle QTc > 500 ms chez 12 patients (1,3 %). 58 patients (6,3 %) ont présenté un allongement de l'intervalle QTc > 60 ms par rapport à la valeur de référence. Une analyse de tendance centrale des données d'intervalle QTc à des concentrations moyennes à l'état d'équilibre issues de l'étude A2301, a montré que la borne supérieure de l'IC bilatéral à 90 % pour l'allongement intervalle QTc était de 15,3 ms avec 750 mg de céritinib pris à jeun par rapport à l'inclusion. Une analyse pharmacocinétique suggère que le céritinib entraine un allongement de l'intervalle QTc dépendant de la concentration (voir rubrique Mises en garde spéciales et précautions d'emploi).
Zykadia a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines.
Une prudence particulière s'impose lors de la conduite de véhicules ou de l'utilisation de machines, car les patients peuvent ressentir de la fatigue ou présenter des troubles visuels au cours du traitement.
Les études de pharmacologie de sécurité indiquent que le céritinib est peu susceptible de perturber les fonctions vitales du système respiratoire et du système nerveux central. Les données in vitro montrent que la CI50 pour l'effet inhibiteur du céritinib sur le canal potassique hERG était de 0,4 µM. Une étude de télémétrie in vivo réalisée sur 4 singes a mis en évidence un faible allongement de l'intervalle QT chez l'un des animaux après administration de la plus forte dose de céritinib. Les études d'ECG réalisées chez le singe après 4 ou 13 semaines d'administration de céritinib n'ont pas montré d'allongement de l'intervalle QT ni de tracés ECG anormaux.
Le test du micronoyau réalisé sur des cellules TK6 était positif. Aucun signe de mutagénicité ou de clastogénicité n'a été observé au cours des autres études de génotoxicité in vitro et in vivo menées avec le céritinib. Par conséquent, aucun risque génotoxique n'est attendu chez l'Homme.
Aucune étude de carcinogénicité n'a été réalisée avec le céritinib.
Les études de reprotoxicité (c'est-à-dire les études portant sur le développement embryo-fœtal) réalisées chez la rate et la lapine gravides n'ont pas mis en évidence d'effet fœtotoxique ou tératogène après administration de céritinib pendant l'organogenèse. Toutefois, l'exposition plasmatique de la mère était inférieure à celle observée à la dose recommandée chez l'Homme. Aucune étude non clinique formelle portant sur les effets potentiels du céritinib sur la fertilité n'a été réalisée.
La principale toxicité liée à l'administration de céritinib chez le rat et le singe a été une inflammation des voies biliaires extra-hépatiques accompagnée d'une augmentation du nombre de neutrophiles dans le sang périphérique. À des doses plus élevées, l'inflammation mixte cellulaire et neutrophilique des voies biliaires extra-hépatiques s'est propagée au pancréas et/ou au duodénum. Une toxicité gastro- intestinale a été observée chez les deux espèces, caractérisée par une perte de poids, une diminution de la prise alimentaire, des vomissements (chez le singe), des diarrhées et, à des doses élevées, par des lésions histopathologiques comprenant une érosion, une inflammation des muqueuses et la présence de macrophages spumeux dans les cryptes et la sous-muqueuse duodénales. Une atteinte hépatique a également été observée chez les deux espèces, à des expositions à peu près équivalentes à l'exposition clinique obtenue à la dose recommandée chez l'Homme, avec notamment une élévation minime des transaminases hépatiques chez quelques animaux et une vacuolisation de l'épithélium des voies biliaires intra-hépatiques. Des macrophages alvéolaires spumeux (phospholipidose confirmée) ont été observés dans les poumons des rats mais pas des singes, et les ganglions lymphatiques des rats et des singes présentaient des amas de macrophages. Les organes cibles ont montré une récupération partielle à complète.
Des effets sur la thyroïde ont été observés chez le rat (légères augmentations des concentrations de thyréostimuline et de triiodothyronine/thyroxine T3/T4 sans corrélation microscopique) et chez le singe (déplétion de la colloïde chez les mâles dans une étude de 4 semaines et hyperplasie diffuse des cellules folliculaires avec augmentation de la thyréostimuline chez un singe sous haute dose dans une étude de 13 semaines). Dans la mesure où ces effets non cliniques ont été légers, variables et contradictoires, la relation entre le céritinib et les changements survenus dans la glande thyroïde chez
les animaux est incertaine.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
liste I.
Médicament nécessitant une surveillance particulière pendant le traitement.
Médicament soumis à prescription hospitalière, réservé aux spécialistes et services en CANCEROLOGIE ou ONCOLOGIE MEDICALE.
Gélule
Gélule composée d'un corps blanc opaque et d'une coiffe bleue opaque, de taille 00 (longueur approximative : 23,3 mm), portant l'inscription « LDK 150MG » imprimée sur la coiffe et « NVR » sur le corps de la gélule et contenant une poudre blanche à presque blanche.
Plaquettes thermoformées en PVC/PCTFE (polyvinylchloride/polychlorotrifluoroéthylène) - Aluminium contenant 10 gélules.
Conditionnements contenant 150 (3 boîtes de 50) gélules.
Chaque gélule contient 150 mg de céritinib.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Contenu de la gélule
Cellulose, microcristalline Hydroxypropylcellulose, faiblement substituée Glycolate sodique d'amidon (type A)
Stéarate de magnésium Silice colloïdale anhydre
Enveloppe de la gélule
Gélatine Indigotine (E132)
Dioxyde de titane (E171) Encre d'impression
Vernis à la gomme laque (blanchie, déparaffinée) 45 % Oxyde de fer noir (E172)
Propylène glycol
Hydroxyde d'ammonium 28 %